小社が立地する熊本にある熊本大学とパートナーシップ協定を結んでいるタイの大学研究者によるライフサイエンス分野の連載(4 回)をお届けします。
Targeting DNA Repair Mechanisms: Exploiting Acquired Vulnerabilities to Overcome Resistance in Cholangiocarcinoma

Donniphat Dejsuphong
Program in Translational Medicine,
Chakri Naruebodindra Medical Institute,
Faculty of Medicine Ramathibodi Hospital,
Mahidol University.

Rakkreat Wikiniyadhanee
Program in Translational Medicine,
Chakri Naruebodindra Medical Institute,
Faculty of Medicine Ramathibodi Hospital,
Mahidol University.

Tassanee Lerksuthirat
Research Center,
Faculty of Medicine Ramathibodi Hospital,
Mahidol University.

Sermsiri Chitphuk
Offices of Health Science Research,
Faculty of Medicine Ramathibodi Hospital,
Mahidol University.
Abstract
Current therapeutic strategies for Cholangiocarcinoma (CCA) are frequently constrained by the rapid onset of chemoresistance, rendering standard-of-care regimens like gemcitabine and cisplatin (GEM/CIS) ineffective over time. Recent advances in genomic profiling have revealed that CCA possesses a high frequency of alterations in DNA Damage Response (DDR) genes, providing a landscape for targeted intervention. This article explores the transition from traditional cytotoxic approaches to the strategic inhibition of DNA repair enzymes, such as PARP and ATR. We argue that the adaptive reprogramming of CCA cells during treatment creates specific acquired vulnerabilities — molecular dependencies that do not exist in drug-naïve cells. By mapping these resistance trajectories, our research demonstrates that combinations of PARP and ATR inhibitors can effectively exploit the genomic instability inherent in CCA. This approach transforms the challenge of drug resistance into a therapeutic opportunity, offering a blueprint for more durable and personalized clinical interventions.
The Crisis of Resistance in Cholangiocarcinoma
Cholangiocarcinoma (CCA) represents a heterogeneous group of malignancies arising from the biliary epithelia 1). Despite its relatively low global incidence compared to other gastrointestinal cancers, its mortality rate remains alarmingly high, largely due to late-stage diagnosis and a lack of effective long-term therapies. For decades, the combination of gemcitabine and cisplatin (GEM/CIS) has remained the frontline standard; however, the median progression-free survival remains measured in months rather than years.
The primary obstacle to clinical success is the inherent and acquired resistance to DNA-damaging agents. CCA tumors often exhibit a “plastic” phenotype, allowing them to circumvent the DNA lesions induced by cisplatin through the upregulation of bypass repair mechanisms. Recent large-scale genomic analyses have identified that approximately 20–30% of CCA cases harbor mutations or epigenetic silencing in DDR-related genes, including BAP1, ARID1A, and PBRM1 2), 3). This genomic landscape suggests that CCA cells may be “prime” for a collapse in genomic integrity if their compensatory repair pathways are strategically blocked.
Mechanism of Targeting DNA Repair
The maintenance of genomic stability is governed by a complex, coordinated network of DNA repair pathways, collectively termed the DNA Damage Response (DDR). In malignant cells, these pathways are often co-opted or selectively inactivated, creating unique therapeutic vulnerabilities relatively specific to cancer cells 4).
- PARP Inhibitors and the “Trapping” Phenomenon
PARP1 is an essential enzyme for the detection and repair of single-strand breaks (SSBs) via the Base Excision Repair (BER) pathway. When PARP1 is inhibited, SSBs persist and are converted into lethal double-strand breaks (DSBs) during the S-phase of the cell cycle when they encounter replication forks 5).
Modern PARP inhibitors (PARPi) such as olaparib, rucaparib, and talazoparib act not only through catalytic inhibition but also through “PARP trapping” 6). This mechanism involves the stabilization of PARP-DNA complexes, which act as physical barriers to replication machinery. In cells with Homologous Recombination (HR) deficiency — such as those with BRCA1/2 mutations — these DSBs cannot be repaired accurately, leading to “synthetic lethality.” Among clinical inhibitors, talazoparib is identified as a significantly more potent trapper, showing higher cytotoxic potency in various models. - The ATR/ATM Axis: Master Regulators of DDR
While ATM (Ataxia-Telangiectasia Mutated) is the primary transducer for DSBs, ATR (Ataxia Telangiectasia and Rad3-related) is the apical kinase responding to replication stress and SSBs. In the context of cancer, ATR is often upregulated to protect the cell from the high levels of replication stress induced by oncogenes or exogenous DNA damage 7), 8).- ATM Function: ATM responds primarily to DSBs by phosphorylating targets like H2AX (creating γH2AX) and recruiting high-fidelity repair machinery.
- ATR Function: ATR stabilizes stalled replication forks, preventing their collapse into DSBs. By inhibiting ATR, we force the cell to continue through the cell cycle with damaged or incomplete DNA. This leads to “mitotic catastrophe”, where the cell attempts to divide with fragmented chromosomes.
- NHEJ Inhibitors: Modulating Error-Prone Repair
Non-homologous end joining (NHEJ) is a predominant, yet error-prone, repair pathway for DSBs that operates throughout the cell cycle. Research using biosensors, such as 53BP1 fluorescent tagging, has identified compounds that modulate this pathway. By inhibiting NHEJ, we can sensitize tumors that have become reliant on this “quick-fix” pathway when their high-fidelity HR pathways are compromised or therapeutically blocked 9).
Current Usage and Limitations in Clinical Practice
While DNA repair inhibitors have significantly advanced oncology, their application in solid tumors faces substantial hurdles that necessitate new combinatorial strategies.
- Breast Cancer: PARPi are approved for BRCA-mutated cases, but acquired resistance frequently emerges through “reversion mutations” that restore the BRCA reading frame or the restoration of HR activity via the loss of end-resection antagonists 10), 11).
- Pancreatic Cancer: Despite clinical benefits in BRCA-mutated metastatic cases, the aggressive nature of the disease and the dense stroma often lead to the rapid bypass of repair protein dependency 12).
- Prostate Cancer: While effective in metastatic castration-resistant models (mCRPC) harboring DDR mutations, tumor heterogeneity necessitates the identification of robust biomarkers beyond simple mutation status to predict long-term responses 13).
The Concept of Acquired Vulnerability in DNA Repair
Rather than focusing solely on primary oncogenic drivers, we investigate “acquired vulnerability”. When CCA cells are exposed to primary therapeutic agents, they undergo molecular reprogramming to facilitate survival 14). In doing so, they manifest a fundamental biological trade-off by developing new weaknesses — specifically, a heightened dependency on alternative DNA repair pathways or stress response axes 15).
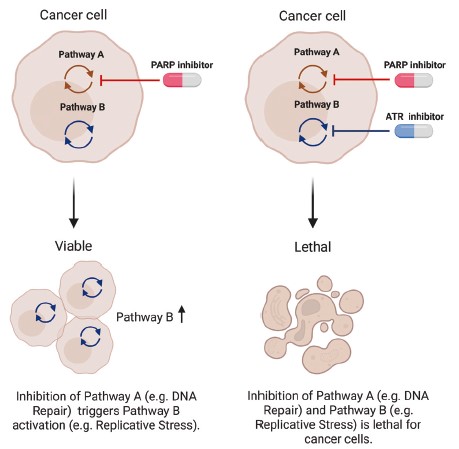
Drug-naïve cells develop resistance to "Drug A" (e.g., a PARP inhibitor) by upregulating compensatory repair pathways or adaptive responses to replicative stress (e.g., the ATR/CHK1 axis). This adaptation creates a novel, targetable dependency for "Drug B" (an ATR inhibitor).
Finding the Right Combination: PARP + ATR Inhibition
A key insight from our research is that exploiting these vulnerabilities is most effective when secondary treatments are combined with primary therapy to induce a concerted multifaceted disruption of the cancer cell's genomic integrity 10), 16)-18).
Synergy of PARP and ATR Inhibitors
Our laboratory has demonstrated that the combination of PARP and ATR inhibitors (e.g., olaparib and AZD6738) provides a powerful synergistic effect. In CCA cell lines and TK6 models, we observed that ATR inhibition prevents the compensatory survival mechanisms that cells use to bypass PARP inhibition 19), 20).
This combination results in:
- Increased Replication Stress: Simultaneous inhibition leads to an accumulation of unrepaired DNA lesions.
- Exhaustion of Repair Capacity: The cell is forced into a “checkpoint collapse”. ATR inhibition prevents the G2/M arrest that would normally allow the cell to repair damage. Instead, the cell enters mitosis prematurely, leading to apoptosis even in cells that were previously resistant to PARPi monotherapy.
- Genomic Instability: We observed significant increases in micronuclei formation and γH2AX foci in treated cells, markers of catastrophic DNA fragmentation.
Validation and Future Directions
For clinical translation, we need to validate these findings using patient-derived organoids (PDOs), capture the unique heterogeneity of CCA more accurately than traditional 2D cultures. However, several challenges remain:
- Biomarker Development
- Functional HRD Assays: Measuring the actual repair capacity of the tumor rather than just the presence of mutations.
- Liquid Biopsies: Monitoring circulating tumor DNA (ctDNA) to track the “reversion” of phenotypes or the emergence of new DDR mutations in real-time.
- Scheduling and Toxicity Management
- Determining whether sequential or concomitant administration of DDR inhibitors best prevents resistance is critical. Because both PARPi and ATRi can cause myelosuppression, research is focused on “pulsatile” dosing schedules that allow the bone marrow to recover while maintaining selective pressure on the tumor.
Conclusion
We redefine drug resistance in CCA not as an insurmountable barrier, but as a window of opportunity. By targeting specific DNA repair adaptations — such as the dependency on the replicative stress following PARP inhibition — we can design sophisticated, adaptive combination therapies. This paradigm shift from static genetic targeting to dynamic adaptation targeting holds the potential to significantly improve survival outcomes for patients with aggressive malignancies like CCA.
[Contact]
Donniphat Dejsuphong (MD, PhD)
ORCID: 0000-0001-8367-9415 (Associate Professor)
Program in Translational Medicine, Chakri Naruebodindra Medical Institute, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Samut Prakan 10540, Thailand
Current research area: DNA repair and diseases from mutation, hereditary cancer syndromes, genetic testing and biological indicators
Rakkreat Wikiniyadhanee (PhD)
ORCID: 0000-0002-2886-5415 (Postdoctoral Researcher)
Program in Translational Medicine, Chakri Naruebodindra Medical Institute, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Samut Prakan 10540, Thailand
Current research area: CRISPR-Cas9 homology-directed repair (HDR) editing, PARP inhibitor combination therapy
Tassanee Lerksuthirat (PhD)
ORCID: 0000-0001-9526-951X (Senior Researcher)
Research Center, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand
Current research area: Functional genomics in DNA damage and repair pathways, drug discovery, preventive and social medicine
Sermsiri Chitphuk (MS)
ORCID: 0000-0002-8149-0341 (Medical Technologist)
Offices of Health Science Research, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok 10400, Thailand
Current research area: DNA repair and diseases from mutation, hereditary cancer syndromes, genetic testing and biological indicators