計算化学に基づく色素分子の励起状態の理論解析
Computational Chemistry for Theoretical Analysis of Excited States in Dye Molecules

柳井 毅
名古屋大学
トランスフォーマティブ生命分子研究所
教授

藤本 和宏
名古屋大学
トランスフォーマティブ生命分子研究所
特任准教授

稲井 直人
名古屋大学大学院
理学研究科 理学専攻
助教
Abstract The expanding applications of molecular dyes in fluorescence imaging, OLEDs, and photocatalysis demand theoretical tools capable of reliably predicting absorption and emission spectra and excited-state dynamics. This review outlines current computational approaches to dye photophysics, focusing on quantum chemical calculations based on density functional theory (DFT) and its time-dependent extension (TD-DFT) for analyzing excited states and transition processes. We showcase our recent research on fluorescence quantum yields of phosphorus-bridged stilbenes, energy-transfer analysis of photosynthetic antenna complexes, in silico exploration of novel π-frameworks, and spin-orbit-coupling-driven dynamics simulations. These examples demonstrate how quantum chemical calculations can quantitatively uncover mechanisms inaccessible to experiment, advancing theory-experiment interplay in dye-molecule research.
1.はじめに
蛍光イメージングプローブや有機 EL 材料、光触媒など、色素分子の応用範囲は急速に広がっており、それに伴い、分子設計段階から光吸収・発光特性や失活機構を予測することの重要性が高まっている。本稿では、計算化学手法を用いた色素分子(主には有機分子)の励起状態解析の現状と展望を概説する。計算機性能と量子化学ソフトウェアの発展により、基底状態の最適化構造だけでなく、励起エネルギーや遷移状態の分子構造の特定が計算機上でルーチンに実施されている。さらには、状態間遷移の速度定数やエネルギー・電子移動のダイナミクスなど、量子的な過程の詳細な情報を評価できる時代に入りつつある。本レビューでは、まず量子化学計算の主力である密度汎関数理論(DFT/TD-DFT)を中心に、励起状態を扱う基礎理論を紹介する。続いて、当研究室で行った理論研究として、架橋スチルベン類縁体の蛍光量子収率の計算、光合成アンテナのような複合系、新奇骨格の in silico 探索、スピン軌道相互作用を含むダイナミクス計算など、色素分子研究における計算化学の役割がどのように展開されているかを示す。特に、本稿で取り上げる例はいずれも、実験的知見だけでは捉えにくい励起状態の詳細を、量子化学計算・電子状態理論に基づいて、高精度に明らかにしたものであり、計算と実験の協奏による色素分子の化学研究の将来像を示すことを目指したい。
2.励起状態を扱う量子化学計算法
2.1 量子化学計算とは
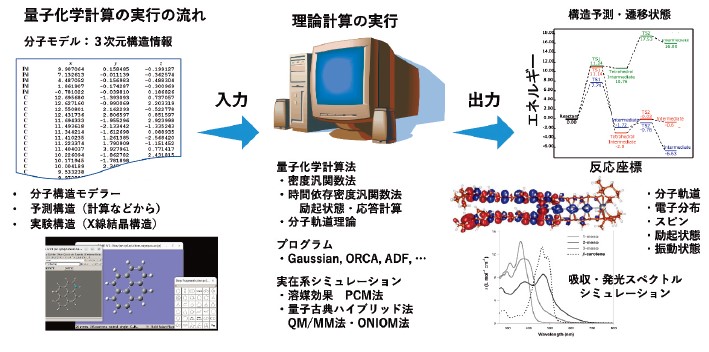
量子化学計算は、分子の電子状態を量子力学に基づいて記述し、分子構造や化学反応を数値的に予測するための理論的手法である1, 2)。実験では観測が難しい短寿命の中間体や遷移状態を “計算機上で可視化できる” 点は大きな利点であり、実験化学を補完する研究手段として広く利用されている。近年は高速計算機の普及やソフトウェア環境の整備3)により、理論の専門家以外の化学者が利用する場面が一般化している。
分子の性質を記述する際の中心となるのが、電子の振る舞いを定めるシュレディンガー方程式である。この方程式を厳密に解くことは多くの場合不可能であるため、量子化学では多様な近似法や計算アルゴリズムが発展してきた。その中でも密度汎関数法(DFT)は、計算精度と計算コストのバランスが良く、分子軌道やエネルギーを効率的に求められる方法として現在最も広く使われている。DFT を含む量子化学手法の発展は、ポープルやコーンらによる基礎理論の構築に支えられており、ノーベル化学賞に結実したことでもよく知られている。
実際の計算では、ユーザーは分子の三次元構造を入力するだけで、経験的パラメータに頼ることなく、エネルギーや分子軌道、電子密度といった量子論的情報を得ることができる。量子化学計算の強みとして特に重要な点は次の二つである。(1)電子の波動関数に基づき化学結合を直接記述できるため、反応経路やポテンシャルエネルギー面を解析できること、(2)基底状態だけでなく励起状態のエネルギーや性質を扱えるため、光吸収・発光や電子移動など分子の電子物性に関わる現象を解析できることである。さらに、単一(気相中)の分子だけでなく、溶媒の効果を取り入れる連続誘電体モデル(PCM)や、タンパク質環境などを扱う QM/MM 法など、より現実の条件に近いシミュレーション手法も確立されている。計算機性能の向上とソフトウェアの進展により、現在では理論化学の専門家でなくともルーチンな DFT計算を実行でき、構造情報さえあれば高度な電子状態解析を容易に行える環境が整いつつある。
2.2 密度汎関数理論と励起状態理論
密度汎関数法(DFT)は、分子から固体材料まで多様な系の電子状態を扱う第一原理計算法であり、現代量子化学の主要手法となっている2)。電子密度を基本変数とすることで、全電子の波動関数を直接扱う必要がなく、エネルギーや構造、反応性を効率よく求められる点が特徴である。電子密度と基底状態エネルギーの対応関係がコーンらの定理によって保証され、DFT が厳密な量子力学に基づく理論であることが示された。分子向け近似やアルゴリズムの整備により、現在では構造最適化や反応経路解析に欠かせない標準手法となっている。実用的な DFT の中心はコーン– シャム法である。電子密度のみでは運動エネルギーを十分に表せないため、非相互作用電子系の軌道を導入し、残る電子間相互作用を「交換相関汎関数」として近似する。局所密度近似や密度勾配補正、さらにハイブリッド汎関数(B3LYP など)が広く利用され、計算精度と効率のバランスに優れている。
励起状態を扱うためには、時間依存密度汎関数法(TD-DFT)が用いられる。時間変化する電子密度への線形応答を解析することで、電子励起エネルギーや吸収・発光波長を求める手法であり、多くの量子化学プログラムに実装されている。バレンス(価)電子励起については良好な精度を示し、色素分子の光吸収特性解析に有効である。一方、電子の空間的な転送を伴う電荷移動励起では、従来の汎関数が遠距離ポテンシャルを正確に再現できず、励起エネルギーを過小評価する問題があった。それに対応して、長距離補正型ハイブリッド汎関数の開発による改善が達成され2, 4)、TD-DFT 法は色素分子の励起状態計算において重要性を増している。
2.3 状態遷移・エネルギー移動の計算法
有機色素分子の光物性は、励起後にどの経路で失活(輻射・無輻射)あるいは反応へと進むのかを決定する電子状態遷移の理解に大きく依存しており、その解析にはシミュレーション計算が重要な役割を担う。近年では、量子化学計算と組み合わせることで、遷移速度定数を理論的に評価する試みが現実的になってきた5)。輻射失活、内部転換、項間交差などの過程は、時間依存シュレディンガー方程式に対する摂動論、すなわちフェルミの黄金律に基づき、遷移双極子モーメント、非断熱結合、スピン– 軌道相互作用、さらには振動状態の重なり積分を入力として速度定数が求められる。これらの計算手法は、熱活性化遅延蛍光(TADF)分子6)の逆項間交差の高速化設計や、光触媒の反応機構解析など、多様な物性設計研究に応用されている。一方で、円錐交差を経由した遷移は、励起状態上の反応座標に沿ったエネルギープロファイルから活性化エネルギーを評価し、アイリングの式などを用いて速度論的推定を行うアプローチが補完的に用いられる7, 8)。
分子間のエネルギー移動の理解は、状態遷移解析と並ぶ重要な理論計算の対象である。代表例として FRET(蛍光共鳴エネルギー移動)は、色素間のエネルギー輸送を高感度に反映する現象として知られ、分子間距離の指標として広く利用されている。理論的には、遷移双極子間のクーロン相互作用に基づくフェルスター理論が基盤となり、蛍光・吸収スペクトルの重なり積分や配向因子を用いてエネルギー移動速度を定量できる。これらの物理量を量子化学計算から求めることが可能となっている。
また、光合成アンテナ複合体や分子凝集体のように多数の色素が密接に相互作用する系では、単一の二分子近似では記述しきれない集団励起効果が重要となる。この場合、各色素分子の励起エネルギーと分子間電子カップリングを要素として励起子ハミルトニアンを構築し、その対角化によって得られる励起子状態を基にエネルギー移動経路や光吸収特性を解析する励起子理論が有効である9-11) 。分子間電子カップリングの計算法としては、モノマーの電荷分布から相互作用を高精度に再現する遷移電荷相互作用(TrESP)法12)が広く用いられており、低計算コストで大規模系のエネルギー移動解析を可能とする点で特に有用である。
3. 色素分子の励起状態解析への計算化学の応用
3.1 リンの酸化状態がリン架橋スチルベン類縁体の蛍光量子収率にもたらす影響
新規の物性を持つπ共役有機分子を開発する手段の1つとして、典型元素をπ共役系に組み込む骨格開発が行われている。中でも、リンは注目されている典型元素の1つであり、高い光耐久性を持つ分子や近赤外領域に発光を示す分子など、種々の分子開発に用いられてきた。リン含有有機色素の特徴の1つに、リン原子の状態による顕著な電子状態の変化があげられる。3 価のリンが非共有電子対を持つ状態、ルイス酸に配位した状態、酸化によって5 価になった状態、と状態に応じた物性を示すとされている 13)。山口らは、リン架橋スチルベン類縁体が、架橋するリンの酸化状態や主骨格の種類によって大きく異なる蛍光波長を示すことを報告した14)。同時に、蛍光量子収率もまたリンの酸化状態などに大きく依存することが判明した。しかしながら、同じ酸化であっても収率が向上するものと低下するものの両方が存在し、リンの酸化状態の違いによる量子収率変化の要因を理解するには至っていなかった。
この問題に対して我々は、一重項最低励起(S1)状態からの状態間遷移(図2a)––– 蛍光発光、内部転換、項間交差––– それぞれの速度定数を計算することにより、各分子の主要な失活過程及び蛍光量子収率を調べた15)。TD-DFT に基づき、合成された分子(P-Ben とPO-Ben)とモデル分子(P-Thio とPO-Thio)のS1状態と三重項励起状態を計算した。得られた構造を用いたTVCF理論5)に基づく速度定数計算は、分子の違いによる蛍光量子収率の変化を定性的に再現した(図2b)。分子P-Benには S1状態より僅かに安定な三重項励起状態が2つ存在し、それらへの項間交差が主要な過程になるために蛍光量子収率の低下を引き起こすと計算された。ここへリンの酸化またはチオフェンの導入を行うと、項間交差が抑制されて量子収率が大幅に向上すると計算された。これは、部分構造の変化によって最高占有軌道(HOMO)や最低非占有軌道(LUMO)の準位が変化し、HOMOーLUMO励起に帰属されるS1状態が安定化され、結果として三重項状態が相対的に不安定化したことによると示唆された。一方、リンの酸化とチオフェンの導入の両方を行った分子PO-ThioではS1状態が強く安定化され、結果として内部転換が支配的になり、量子収率の低下を招くと計算された。このように、リンの酸化状態の変化がS1状態のエネルギー準位を変化させ、項間交差の抑制や内部転換の加速を引き起こすことが、リン架橋スチルベン類縁体の蛍光量子収率の変化をもたらす物理化学的要因である可能性を示した。
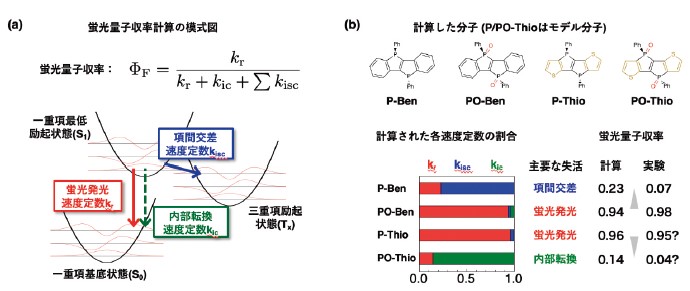
(a)蛍光量子収率の計算式と関係する速度定数。(b)計算した分子と計算された速度定数の割合および蛍光量子収率。
3.2 珪藻の光合成アンテナの特異な光学機能の量子化学計算
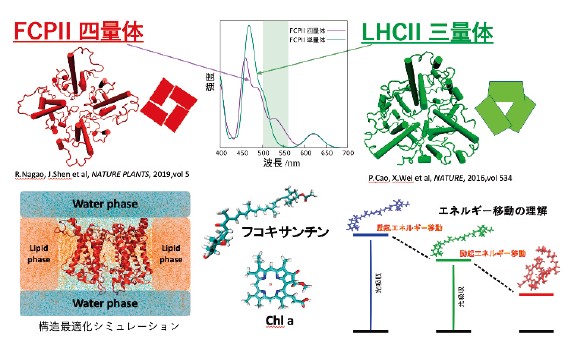
光合成アンテナは多数の色素分子が集積して光エネルギーを吸収・移動する巨大複合体であり、その吸収特性は生物種によって大きく異なる。特に珪藻が示す赤褐色の体色は 500–560 nm 領域の強い吸収に起因するが、その分子機構は未解明であった。本研究では、クライオ電子顕微鏡で明らかになった FCPII の構造を基に、量子化学計算と励起子理論を統合した計算法を用いて、特異な光吸収の起源を分子レベルで解析した9)。
励起子理論では、各色素の励起エネルギー・遷移双極子モーメント・分子間電子カップリングを入力として励起子ハミルトニアンを構築し、その対角化から複合体全体の吸収スペクトルを求める。FCPII は数十個の色素を含み、全電子励起計算は実質的に不可能であるが、励起子モデルを用いることで大規模系にみられる相互作用を効率的に取り込める。本研究では特に、電子カップリングにTrESP 法を導入し、カロテノイドとクロロフィルが密集した環境における相互作用を高精度に評価した。
計算によって得られた FCPII 四量体の吸収スペクトルは実験結果を良好に再現し、500–560 nm の吸収帯が確かに存在することが確認された。スペクトル分解の結果、この吸収は通常のフコキサンチンとは異なる配置に置かれた特別な分子、フコキサンチン-S に由来することが明らかになった。立体構造比較から、この位置にはホウレンソウ LHCII には対応するカロテノイドが存在せず、フコキサンチン-S が珪藻アンテナに特有の構造要素であることが示唆された。
さらに励起子解析により、四量体化に伴うプロトマー間の近接化がフコキサンチン-S と荷電アミノ酸や Chl a の静電相互作用を強め、励起エネルギーを低下させて赤方吸収を生じることが判明した。また電子カップリングの計算から、フコキサンチン-Sは他のフコキサンチンよりも効率的に Chl a へエネルギー移動を行い、補助的な集光素子として機能していることが示された。
本研究は、励起子理論と量子化学計算を組み合わせることで、光合成アンテナのような大規模色素集合体の光物性を高精度に記述できることを示し、珪藻が海中環境に適応する分子戦略を包括的に理解する手がかりを与えた。
3.3 色素分子のin silico探索に向けた量子化学アルゴリズムの高度化
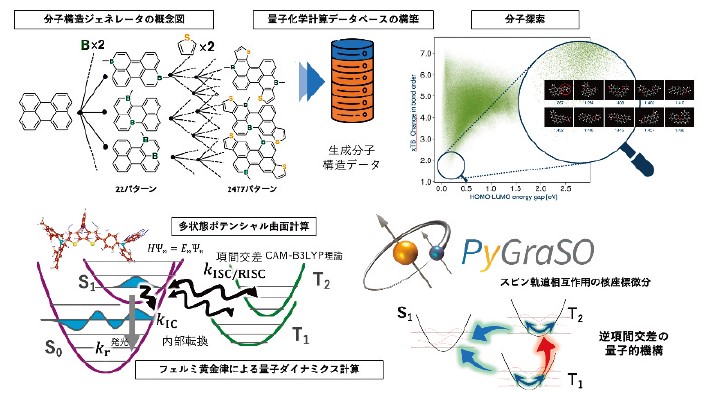
近赤外発光色素や TADF 分子など、高度な光機能をもつ色素の設計には、膨大な化学空間の中から有望候補を効率よく絞り込む in silico 探索が不可欠になりつつある。本研究では、分子構造ジェネレータ16)と量子化学計算に加え、スピン軌道相互作用を精密に扱う励起状態ダイナミクス理論17)を組み合わせることで、色素分子探索のための計算枠組みを高度化し、従来は経験的試行錯誤に依存していた分子設計を理論主導へと転換することを目指している。
近年我々は、多環芳香族炭化水素(PAH)骨格にホウ素ドープとチオフェン縮環を施すという設計コンセプトに基づき、独自に開発した分子構造ジェネレータにより約2500 個の候補構造を自動生成し、TD-DFT 計算によって吸収・発光波長と遷移双極子モーメントを一括評価した16)。その結果、近赤外領域で強い蛍光を示す候補を抽出し、実際に合成した標的分子が高い蛍光量子収率を示すことを確認した。設計原理で定めた化学空間を網羅的にスクリーニングし、計算結果を実験で検証するというスキームは、新規π共役骨格の探索や機能性ナノグラフェンの開発に有効な戦略であり、今後の高スループット分子探索への展開が期待される。
さらに、スピン反転を伴う無輻射失活過程(項間交差・逆項間交差)を扱うため、その量子的な過程を支配するスピン軌道相互作用を高精度に評価するソフトウェア開発も進めている。 PyGraSO18)はその一例であり、スピン– 振動結合項を解析的な核微分により高精度かつ効率良く取り込むことを可能にするプログラムである。このプログラム開発により、多数の TADF 分子やフォトレドックス触媒候補に対する RISC・ISC 速度定数の高精度予測が現実的となった。これにより、近赤外発光分子のような放射・無輻射過程が競合する系に対しても、発光波長だけでなく寿命や量子収率を同時に最適化するための in silico 設計指針を与えうる計算基盤が整備されつつある。今後は、機械学習的な分子生成モデルや実験データとの連成解析と組み合わせることで、色素材料設計における理論駆動型探索の一層の加速と、真に新規な分子骨格の創出が期待される。
4. おわりに
本稿では、密度汎関数理論に基づく基礎的な励起状態計算から、状態遷移速度やエネルギー移動の理論評価、さらには光合成アンテナや近赤外発光分子を対象とした大規模シミュレーションまで、計算化学が色素分子研究にもたらしている知見を概観した。これらの例は、量子化学計算が単なるスペクトルの再現にとどまらず、無輻射失活過程の詳細な解析や電子カップリング・スピン軌道相互作用の定量化を通じて、光機能発現の分子設計指針を与える段階に達していることを示している。今後は、機械学習ポテンシャルや自動分子生成アルゴリズムとの連携、高度なダイナミクス計算法の普及により、より複雑な環境下での励起状態挙動を現実的なコストで追跡できるようになるだろう。計算化学に基づく励起状態解析は、実験ではアクセスが難しい情報を補完しつつ、新しい色素材料や光機能の開拓を先導する中核的な役割を担うと期待される。そのためには、計算結果の不確かさや理論の適用限界を意識しつつ、実験とのフィードバックを通じてモデルを高度化していく相乗性が重要である。本稿が、色素分子の励起状態を先端的計算化学の視点から捉え直し、新たな研究展開を構想する一助となれば幸いである。
| [ 著者プロフィール ] | |
| 氏名 | 柳井 毅(Takeshi Yanai) |
|---|---|
| 所属 |
名古屋大学 トランスフォーマティブ生命分子研究所 〒464-8601 愛知県名古屋市千種区不老町 |
| 出身学校 | 東京大学大学院 |
| 学位 | 博士(工学) |
| 専門分野 | 量子化学・計算化学 |
| 現在の研究テーマ | 量子化学計算基盤の高度化と応用 |
| [ 著者プロフィール ] | |
| 氏名 | 藤本 和宏(Kazuhiro J. Fujimoto) |
|---|---|
| 所属 |
名古屋大学 トランスフォーマティブ生命分子研究所 〒464-8601 愛知県名古屋市千種区不老町 |
| 出身学校 | 京都大学大学院 |
| 学位 | 博士(工学) |
| 専門分野 | 理論化学 |
| 現在の研究テーマ | 生命に潜む量子現象の解明、インシリコスクリーニングによる分子設計 |
| [ 著者プロフィール ] | |
| 氏名 | 稲井 直人(Naoto Inai) |
|---|---|
| 所属 |
名古屋大学大学院 理学研究科 理学専攻 〒464-8602 愛知県名古屋市千種区不老町 |
| 出身学校 | 名古屋大学大学院 |
| 学位 | 博士(理学) |
| 専門分野 | 量子化学 |
| 現在の研究テーマ | 有機色素分子の電子励起状態 |