GPx4研究からみた脂質酸化依存的細胞死
―リポキシトーシスとフェロトーシス―
Lipid peroxidation dependent cell death ― lipoxytosis and ferroptosis ―
from lessons of GPx4 research

今井 浩孝
北里大学
薬学部 衛生化学
教授
Abstract GPx4 is one of the major antioxidant enzymes that reduce phospholipid hydroperoxides in biological membranes in a glutathione-dependent manner. There are three types of GPx4, each regulating localized lipid oxidation in different organelles. Mitochondrial-type GPx4 inhibits apoptosis and ferroptosis. Cytoplasmic GPx4 is essential for cellular and individual survival and inhibits iron-dependent lipid oxidation-induced cell death ferroptosis and iron-independent lipid oxidation-induced cell death lipoxytosis. Nucleolar GPx4 is involved in nucleolar defense. In this review, we outline the different cell death regulatory mechanisms of GPx4 from our GPx4 research.
1. はじめに
GPx4(リン脂質ヒドロペルオキシドグルタチオンペルオキシダーゼ)は、生体膜に生じたリン脂質ヒドロペルオキシドをグルタチオン依存的に還元する酵素である。我々の研究は、1995年GPx4のcDNAクローニングからはじまり1)、2003年GPx4ノックアウトマウスが胚発生致死となることを初めて報告し2)、GPx4やビタミンEによる脂質酸化の抑制が細胞、個体レベルで必須であることを明らかにするなど、30年間、GPx4の機能研究に携わってきた。近年、フェロトーシス誘導剤による鉄依存性脂質酸化を介した細胞死が注目され、その制御因子の同定や疾患に関する研究が進展しているが、GPx4は細胞内の主要な酸化脂質消去のセンサー酵素であり、もともとフェロトーシスにとどまらず、アポトーシスや我々が見出している鉄非依存生の脂質酸化依存的な細胞死(リポキシトーシス)などの制御にも関与する。本総説では、これまで我々が明らかにしてきた3つのタイプのGPx4の機能や脂質酸化により誘導される細胞死の制御の重要ポイントを中心に概説する。
2.GPx4 の特徴
GPx4は、転写開始点の違いにより、異なるエクソンの使いわけにより、ミトコンドリア内腔に移行できるミトコンドリア型(mGPx4)、細胞質や核内に存在する非ミトコンドリア型(cGPx4、海外のグループは細胞質型と呼ぶ場合があるが、核内にも存在する)、核小体型(nGPx4、海外のグループは核型と呼ぶ場合があるが、核内では核小体に局在する)がある(図1) 3)。 3つのGPx4はN末端側のオルガネラ輸送シグナル以外のC末端側はいずれも共通した構造をしている。GPx4は活性中心にセレノシステインを有するセレンタンパク質の一つである。セレノシステインは終止コドン(TGA)でコードされており、システインの硫黄がセレンに置き換わったアミノ酸で21番目のアミノ酸である。また終止コドンをセレノシステインとして翻訳するためには、mRNAの3’側非翻訳領域のSECIS配列(セレノシステイン挿入配列)が必要である。我々は、はじめ3つのタイプのGPx4をラット好塩基球がん細胞(RBL2H3細胞)で高発現株を作成し、3つのタイプのGPx4の機能を解析した4)。非ミトコンドリア型GPx4は、IgE受容体シグナルを抑制したり、5- リポキシゲナーゼの活性化の抑制、シクロオキシゲナーゼ活性化の抑制など、細胞内の局所でおきる脂質酸化反応やシグナル伝達経路を制御できることを報告した5)。またミトコンドリア型GPx4はミトコンドリアを経由するアポトーシスやミトコンドリア内電子伝達系障害を抑制できることを報告した。また核小体型GPx4はアクチノマイシンDやドキソルビシンなどの核小体ストレスを防御し細胞死を抑制できることを報告した。これらの結果は3つのタイプのGPx4がそれぞれのオルガネラで別々の機能をしていること、また、それぞれのタイプは、細胞内の局所の脂質酸化を検出するセンサーとして機能することを示している4)。
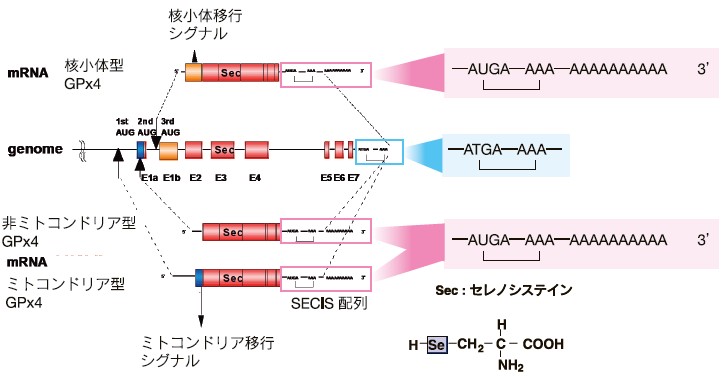
GPx4のゲノム遺伝子は8つのエクソンからなる。E1aエクソンはミトコンドリア移行シグナルを有しミトコンドリア型GPx4(mGPx4)が利用する。E1bエクソンは核小体移行シグナルを有し核小体型GPx4(nGPx4)が利用する。非ミトコンドリア型GPx4(cGPx4)はE1aエクソンの途中から転写がはじまりE1aエクソンの最後のATGを開始コドンとして利用する。E2~E7エクソンは3つのタイプのGPx4に共通に利用される。活性中心のセレノシステイン(Sec)は、微量元素セレンを有するアミノ酸でE3エクソンに終止コドンTGAでコードされる。終止コドンをセレノシステインとして翻訳させるには、mRNAの3'側非翻訳領域のSECIS配列が必要である。
3.3つのタイプのGPxによる細胞死制御
3.1 ミトコンドリア型GPx4によるアポトーシス抑制
ミトコンドリア型GPx4高発現株は、スタウロスポリン、UVや2-デオキシグルコースなどのミトコンドリアを経由するアポトーシスを抑制できることを明らかにした。その抑制メカニズムの解析から、ミトコンドリア型GPx4は、カスパーゼ3の活性化に必要なミトコンドリアからのチトクロームcの放出を抑制できることを示し5)、ミトコンドリア内で生成した活性酸素により、ミトコンドリア特異的なカルジオリピン(CL)が酸化され、カルジオリピンヒドロペルオキシド(CLOOH)になると、カルジオリピンに結合していたチトクロームcが内膜から遊離し、ミトコンドリア外に放出されることを明らかにした6)。また、ミトコンドリア内膜において、チトクロームc放出に関与するミトコンドリアポアの制御に関与するANT(アデニンヌクレオチドトランスロケーター)は、その構造維持やATP輸送にカルジオリピンを必要とするが、脂質二重膜にANTを組み込んだプロテオリポソームを用いたATP輸送実験から、リポソーム内でカルジオリピンヒドロペルオキシド(CLOOH)は、カルジオリピンと競合阻害して、ANTの構造変化によりATP輸送が低下させるが、ホスファチジルコリンヒドロペルオキシド(PCOOH)ではおきない7)。このことから、ミトコンドリア型GPx4は、カルジオリピンの酸化を抑制し、アポトーシスを多面的に抑制できる(図2)。最近、我々は抗がん剤ドキソルビン投与の副作用としての心毒性に、ミトコンドリアにおける鉄依存的な脂質酸化を介したフェロトーシスが関与し、ミトコンドリア型GPx4が抑制する機能をもつことを報告している8,9)。
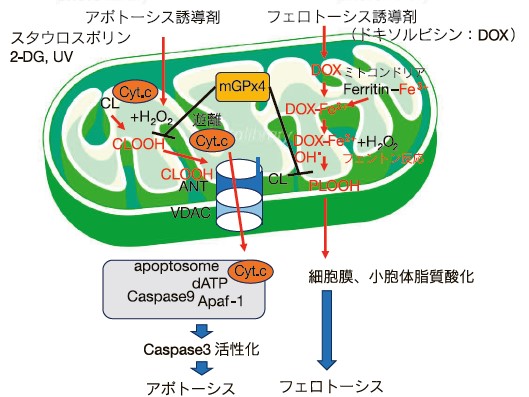
ミトコンドリアを経由するアポトーシス誘導剤は、ミトコンドリア特異的なリン脂質カルジオリピン(CL)を酸化し、カルジオリピンヒドロペルオキシド(CLOOH)を生成する。チトクロームc(Cyt.c)はカルジオリピンに結合しているが、酸化により内膜から遊離する。遊離したCyt.cは、ANT(アデニンヌクレオチドトランスポーター)―VDACからなるPTポアよりミトコンドリア外に放出されると、Apaf-1, dATP, Caspase 9と複合体をつくり、アポプトソームを形成し、カスパーゼ3を活性化してアポトーシスを実行に導く。CLOOHはANTに作用し構造を変化させ、PTポアを開口させる。ミトコンドリア型GPx4はカルジオリピンヒドロペルオキシドを還元することで、チトクロームcのミトコンドリアからの放出を抑制し、アポトーシスを抑制する機能をもつ。また、フェロトーシス誘導剤である抗がん剤ドキソルビシンは、ミトコンドリア内でヘム合成系を低下させ、余剰となった鉄と錯体を形成し、鉄依存的な脂質酸化を誘導し、ミトコンドリアを経由するフェロトーシスを誘導する。ミトコンドリア型GPx4は、ドキソルビシン誘発性のミトコンドリアを介するフェロトーシスを抑制できる。
3.2トランスジェニックレスキュー法を用いたGPx4欠損マウスにおける胚致死のレスキュー解析
2003年我々はGPx4 KOマウスが胚発生過程の7.5日に胚致死となることを明らかにした。交配後、3.5日GPx4欠損受精卵の培養によるInner Cell Mass(ICM)形成系において、WTタイプでは、培養4日後にはHatching後に細胞塊がみられはじめ、培養10日にはICM形成が顕著に観察される。しかし、GPx4 KO受精卵では培養4日目以降細胞塊が形成されない。この実験系にレトロウィルス感染により、3つのタイプのGPx4のcDNAをそれぞれ導入すると、非ミトコンドリア型GPx4のみでICM形成がみられ、ミトコンドリア型GPx4や核小体型GPx4ではみられなかった。またビタミンEを培地に添加することでもGPx4 KO受精卵のICM形成が確認できたが、ビタミンCやMnTBAP、Nアセチルシステインではレスキューできず、ICM形成にはGPx4が必要であるというよりかは、ビタミンEやGPx4による脂質酸化の抑制が必須であることを見つけた。また実際に、このGPx4 KOマウスに、3つのタイプのGPx4の開始コドンATGをそれぞれTTGに変異させたトランスジェニックGPx4マウスゲノム遺伝子を導入したTGマウスと先のGPx4 KOマウスを交配することで、どのタイプのGPx4でマウスの胚致死をレスキューできるのかを明らかにしたところ、非ミトコンドリア型GPx4をKOしたゲノム遺伝子ではレスキューできず、逆にミトコンドリア型GPx4と核小体型GPx4のダブルKOゲノム遺伝子ではレスキューできたことから、胚発生過程ではミトコンドリア型ではなく、非ミトコンドリア型が必須であることが明らかとなった10, 11)。上述したように当初は、ミトコンドリア型GPx4がアポトーシス抑制機能を有することを明らかにしていたので、非ミトコンドリア型GPx4が必須であったことは、当時驚きであった。我々はこのトランスジェニックレスキュー法で、正常のGPx4ゲノム遺伝子をLoxP配列で挟んだTG遺伝子でレスキューしたマウスを、いわゆるfloxマウスとして用い、さまざまな組織特異的なCre recombinase遺伝子を持つマウスと交配することで組織特異的GPx4 KOマウスを作成し、その表現型の解析をおこなっている10,12)。精巣特異的GPx4 KOマウスでは、精母細胞が致死となり、精子数が著しく減少し、さらに精子尾部の折れ曲がりがみられた。実際、我々は精子のGPx4の発現低下した男性不妊症患者を多数見出しており、精子でのGPx4の発現低下は男性不妊症の原因であることを明らかにしている13)。また網膜、心臓、肝臓、軟骨特異的GPx4 KOマウスはいずれもGPx4 KOした組織の細胞は致死となるが、ビタミンE高添加食の投与で致死がレスキューできる。またいずれの組織においても、GPx4のゲノム遺伝子の破壊による組織細胞死は、Cre recombinaseが発現して致死に至る時間は3日以上かかっている。我々はこのGPx4ゲノム破壊によるGPx4 KO細胞死の分子メカニズムを明らかにするために、タモキシフェン誘導型GPx4 KO MEF細胞(ETK1細胞)を作成して、そのGPx4 KO細胞死の分子メカニズムを解析したところ、同様に約3日かかった。またこの細胞死は鉄のキレーター(DFO)では抑制されず、ビタミンEでは抑制された14)。その当時、Conradらのグループが樹立した MEF細胞(Pfa1細胞)では、GPx4 KOによる細胞死が15-LOX依存的、AIF依存的細胞死であると報告しているが14)、我々の樹立した MEF細胞では、15-LOXやAIFをKDしても全く細胞死を抑制しなかった。そこで我々は、このGPx4 KO細胞死の分子メカニズムについて詳細な解析をおこない、リポキシトーシスと名づけた14,16)。
3.3 鉄依存的脂質酸化を介した細胞死(フェロトーシス)
2012年、2014年に米国のStockwellらは、Ras変異がん細胞を殺し、正常細胞は殺さないスクリーニングにより、xCTを阻害するエラスチン17)、GPx4と結合するRSL3を代表とするフェロトーシス誘導剤を見出し18)、エラスチンはグルタチオン量を減少させることにより、RSL3は直接GPx4を阻害することにより、3-7時間でフェントン反応による二価鉄依存的な脂質酸化を誘導し、24時間以内には致死となる細胞死を見出し、フェロトーシスと名づけた(図3)。フェロトーシスは鉄のキレーター(DFO)およびビタミンEや抗酸化剤であるフェロスタチン-1やリプロスタチン-1により、脂質酸化と細胞死が抑制される。GPx4 KOによっても細胞死が誘導されることから、両者は同じ細胞死として考えられて解析されている。フェロトーシスの制御因子の探索研究は、現在、凄まじい勢いで進んでいるが、その分子のほとんどは、脂質の酸化を直接抑制するFSP1などの抗酸化酵素や低分子抗酸化物質の合成制御系や、リン脂質の不飽和度を制御するACSL4等、またGPx4のタンパク質の発現の制御に関わる分子であり、脂質酸化の下流で機能する分子は、細胞膜のチャネルPIEZO1やNINJ1以外にはほとんど明らかではない。フェロトーシスの制御メカニズムは他の総説を参照していただきたい19)。GPx4を単にKOしただけでは、二価鉄は遊離してこない場合もあることから、RSL3やエラスチンでは二価鉄を放出させるメカニズムがフェロトーシスの実行には重要であると考えられる。
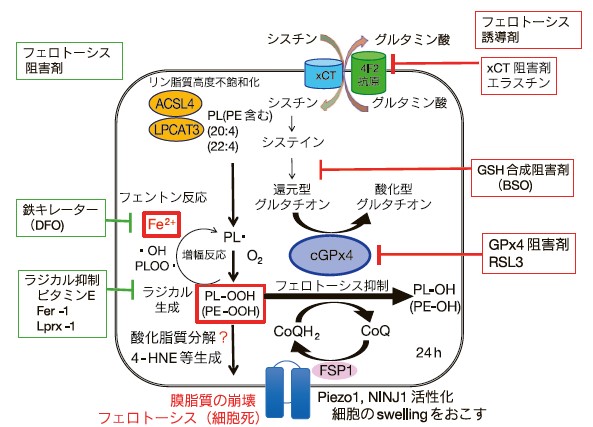
シスチントランスポーター(xCT)の阻害剤エラスチンは、細胞内のグルタチオン(GSH)量を減少させることによりGPx4活性を低下させ、RSL3は直接GPx4と結合することでGPx4を失活させる。一方、エラスチンやRSL3はリソソームから二価鉄を細胞質に放出させ、フェントン反応によりリン脂質特にホスファチジルエタノールアミン(PE)を酸化させる。細胞膜の脂質酸化は、PIEZO1やNINJ1を活性化、高分子化することでチャネルを形成し、Swellingを引き起こした後に膜の崩壊を引き起こす。フェロトーシスは鉄のキレーター(DFO)やラジカル消去剤であるビタミンE、フェロスタチン1(Fer-1)やリプロスタチン1(Lip-1)により抑制できる。cGPx4はビタミンEとともに細胞内の主要なフェロトーシス抑制酵素である。FSP1はユビキノン(CoQ)を還元型にし、脂質酸化を抑制したり、ビタミンEの再生をおこなうフェロトーシス抑制因子である。ACSL4やLPCAT3によるリン脂質のリモデリング経路による膜脂質の不飽和度の亢進はフェロトーシスの感受性を増大させる。
3.4 鉄非依存的な脂質酸化を介した細胞死(リポキシトーシス)
我々が樹立したタモキシフェン誘導型GPx4 KO MEF細胞株(ETK1細胞)では、タモキシフェン添加により、24時間以内にGPx4 KOにともない、ホスファチジルコリン(PC)の酸化体PCOOHの生成がみられ、細胞の増殖がとまり、その後48時間かけて72時間後までに死にいたる。24時間後に見られるこの脂質酸化は、ビタミンEでは抑制できるが、鉄キレーター(DFO)では抑制できないことから、鉄非依存的な脂質酸化を介した細胞死(リポキシトーシス)である14,16)。また脂質酸化の下流の48時間ではERK2のリン酸化が観察された。MEKの阻害剤は、RSL3やエラスチンによるフェロトーシスおよびリポキシトーシスで抑制されたが、MEK1のノックダウン細胞ではリポキシトーシスは抑制されたが、フェロトーシスは抑制されなかった(図4)14)。またERKの阻害剤もリポキシトーシスは抑制されたが、フェロトーシスは全く抑制されない。これらもフェロトーシスとリポキシトーシスは異なるメカニズムである。我々はこれまでに、網羅的shRNAライブラリーによるスクリーニングにより、リポキシトーシス実行因子Lipo-1~5を見出している。Lipo-1はGPx4 KO時の脂質酸化に関与し、Lipo2~5は脂質酸化の下流で機能する分子である。Lipo遺伝子のノックダウンは、リポキシトーシスは抑制できるが、フェロトーシスは抑制できない。このことからもフェロトーシスとリポキシトーシスは異なる。最近、我々は、脂質酸化の下流で機能するLipo KD細胞で細胞死が抑制され、WT細胞で細胞死を誘導できるリポキシトーシス誘導剤を見出した。これらのリポキシトーシス誘導剤による細胞死は48時間かかる。また添加後24時間で脂質酸化が検出されるが、この脂質酸化もビタミンEでは抑制できるが、鉄キレーター(DFO)では抑制されない。また興味深いことにリポキシトーシス誘導剤による脂質酸化は、脂質酸化酵素Lipo-1を介して脂質酸化を誘導する結果が得られてきており、酵素依存的な脂質酸化を介した新規細胞死経路と考えられる。
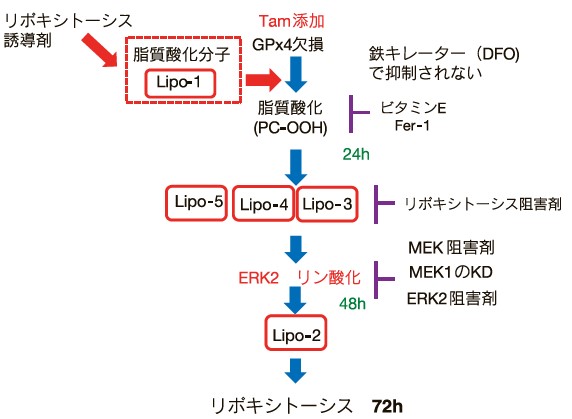
リポキシトーシスは、鉄非依存的な脂質酸化を介した、フェロトーシスとは異なる細胞死で、Swellingはおきない。我々が樹立したETK1細胞では、タモキシフェン(Tam)添加によるGPx4のゲノム遺伝子の破壊は、24時間後にリン脂質のうちホスファチジルコリン(PC)をリポキシトーシス実行因子Lipo-1酵素により酸化し、PCOOHを生成する。この脂質酸化は、鉄キレーター(DFO)で抑制されないが、ビタミンEやFer-1では抑制できる。リポキシトーシスでは24時間後の脂質酸化の下流で、Lipo2~5が機能すること、ERK2のリン酸化が起きることが明らかになっており、MEK阻害剤、MEK1 KDおよびERK2阻害剤で抑制された。また特異的なリポキシトーシス阻害剤も見出している。一方、我々が最近見出したリポキシトーシス誘導剤は、Lipo-1依存的に脂質酸化を誘導し、鉄のキレーターにより抑制されず、細胞死を下流のLipo遺伝子を介して誘導する。
4. おわりに
GPx4はオルガネラ局所で酵素的に生成する脂質酸化や、二価鉄を介したフェントン反応によるランダムな脂質酸化のどちらもグルタチオンを用いて還元できる。現在、細胞死研究分野ではフェロトーシスが最も注目されているが、脂質酸化による細胞死はアポトーシスや、リポキシトーシス、さらに好中球におけるネトーシスでもみられ、それぞれ細胞死の分子メカニズムは異なる。今後は、どこで、どのように生成した酸化脂質分子がそれぞれの細胞死実行経路をスイッチオンにできるのかの分子メカニズムの解析が重要であると思われる。また細胞死における脂質酸化の検出、抗酸化剤に抑制の結果だけではこれらの細胞死が区別できないと思われるため、特に病態解析においては、細胞死に鉄依存性あるいは鉄の沈着が観察されるのかを示すことが重要であると思われる8,20)。またこれらの脂質酸化依存的な細胞死の分類が進むことにより、新たな治療薬の開発が期待される。
| [ 著者プロフィール ] | |
| 氏名 | 今井 浩孝 (Hirotaka Imai) |
|---|---|
| 所属 |
北里大学薬学部衛生化学 〒108-8641 東京都港区白金5-9-1 Tel:03-5791-6235 |
| 出身学校 | 東京大学大学院薬学研究科 |
| 学位 | 博士(薬学) |
| 専門分野 | 脂質生化学、分子生物学 |
| 現在の研究テーマ | リポキシトーシスの分子メカニズムの解明、GPx4が関与する病態と新たな機能解析 |