 |
| トップページ > 光変換前駆体法による有機半導体材料の開発 |
 |
||||||||||||
光変換前駆体法による有機半導体材料の開発
|
||||||||||||
 |
山田 容子
奈良先端科学技術大学院大学 教授 |
Summary:
The finding of clean and renewable energy is one of the major scientific and technological challenges in the 21st century. Solar cells are an attractive alternative considering the availability of solar energy. Organic thin-film solar cells prepared by solution process are one of the good candidates for the promising next generation of solar cells. Recently we have developed a photoconvertible diketone precursors of acenes. The precursors can be converted to the corresponding acenes quantitatively in solution and in film by photoirradiation. Here our recent achievements will be presented.
1.はじめに
再生可能エネルギーの需要が高まり、その開発はサイエンス・テクノロジーの両分野において最重要課題の一つである。なかでも太陽エネルギーは半永久的に享受できる大きなエネルギー源であり、太陽電池の開発が世界的規模で進められている。太陽電池を普及させるためには、低価格・大面積・フレキシブルなデバイスの開発が急務である。現在はシリコン太陽電池が主流であるが、次世代太陽電池として、無機材料を用いた化合物太陽電池、量子ドット太陽電池、低分子・高分子材料を用いた有機太陽電池(OSC)などの開発が盛んに研究されている。
有機半導体材料は、OSC だけでなく、有機発光ダイオード(OLED)、有機薄膜トランジスタ(OTFT)、液晶など、様々な分野で利用されている。電荷輸送は分子間を電荷がいかに効率よく移動できるかにかかっており、HOMO と LUMO のエネルギーギャップや、薄膜中での分子の配列が鍵を握る。そのような観点から考えると低分子有機材料の利点は、単一の構造を有しているため高純度に精製可能なこと、合成化学的に構造をチューニングすることで光学特性・ HOMO / LUMO エネルギーレベル・結晶構造・電荷移動度などを自在にコントロールできることにある 1-5)。本稿では、有機化学的視点から低分子有機薄膜太陽電池を概観し、最後に我々の最近の取り組みをご紹介する。
2. 有機薄膜太陽電池の原理
有機太陽電池は、色素増感太陽電池と有機薄膜太陽電池に大別される。ここでは有機薄膜太陽電池に限定してその構造と評価方法、開発の歴史を簡単に解説する。
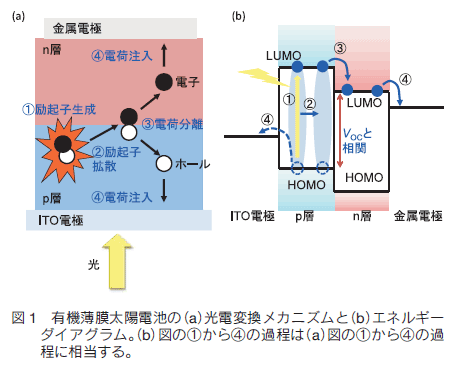
有機薄膜太陽電池の構造を単純化して説明すると、透明半導体電極と金属電極で p 型半導体材料(電子ドナー)と n 型半導体材料(電子アクセプター)からなる活性層を挟み込んだ形をとる。この活性層を光照射すると、
①ドナーまたはアクセプターが励起されて励起子となり、
②励起子が薄膜中を移動してドナーとアクセプターが出会い、
③ドナーからアクセプターへと光励起電子移動が起こり、
④その結果生じたホール(プラスの電荷)は p 層を、電子(マイナスの電荷)は n 層を通ってそれぞれ電極へと送り込まれる仕組みになっている (図 1)。
この 4 つの過程はいずれも極めて重要であり、太陽電池の効率をあげるためには次の 4 つの因子の改良がキーとなる。すなわち
① 光を吸収して励起子が生成する効率
② 生じた励起子が、 p 層と n 層の界面まで移動する効率
③ 励起子からホールと電子への電荷分離が起こる効率
④ 生じたホールと電子が、それぞれ p 層・ n 層を通って両極へ電荷を輸送し、電極へ電荷を注入する効率
である。これらの因子を改良するためには、OSC のデバイス構造はもちろんのこと、ドナー分子・アクセプター分子の HOMO・LUMO レベル、 p 層と n 層から成る活性層の薄膜構造、材料の純度など実に様々な要素が鍵をにぎる。
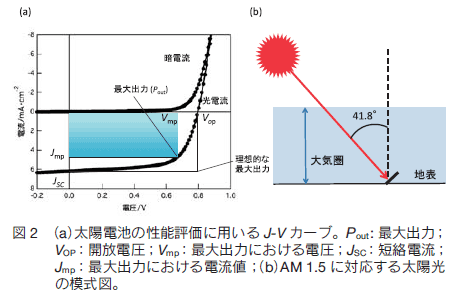
太陽電池の性能は電流-電圧曲線により評価される (図 2a)。光源には AM (エアマス) 1.5 と呼ばれる太陽光スペクトルを照射した標準条件を用いる (図 2b) 。AM 1.5 とは垂直方向から 41.8°傾いた方向から地表に照射される太陽光スペクトルを表す言葉で、太陽が真上にあるとき(AM 1.0)にくらべ大気中を通過する距離が 1.5 倍になり、日本付近の太陽光に相当する。光照射時に電圧を印可しながら電流を測定することで、開放電圧(VOC)、短絡電流密度(J SC)、曲線因子(FF )、エネルギー変換効率(PCE)を求めることができる。VOC は電流が流れないときの電圧に相当し、ドナー分子の HOMO レベルとアクセプター分子の LUMO レベルのエネルギー差と相関がある。J SC はバイアス電圧をかけないときの電流である。活性層の厚みや吸収スペクトルの形状、電荷輸送能、すなわち、薄膜構造に影響される。 FF はその素子の最大出力(Pout)の、理想的な最大出力(VOC J SC)に対する割合であり、次の式で表される。
![]()
光照射により生じた励起子が電極に到達する割合に相当し、1 に近いほど良い特性である。 PCE は得られる電気エネルギー(Pout)の入射光エネルギー(Pin に対する割合であり、次の式が成り立つ。
![]()
この式からわかるように、VOC、J SC、FF のすべての値が大きくなるほど、PCE の値も大きくなる。しかし、それらを同時に満たすことは容易ではない。 VOC の値はドナーの HOMO とアクセプターの LUMO のエネルギー差と相関があるため、ドナー分子とアクセプター分子の分子設計と組み合わせが重要である (図 1b)。
一方 J SC と FF の値は薄膜構造や電極界面の状態が大きく作用する。OSC の実用化には 10 ~ 15% の PCE が必要と言われているが、2011 年には 10 %を超える効率が報告された。そこで次に有機薄膜太陽電池の構造を紹介する。
3. 有機太陽電池の構造
1950 年代に最初に報告された有機薄膜太陽電池は、ショットキー型太陽電池と言われる単層の構造をしており、光合成色素であるクロロフィル a やその類縁体としての Cu フタロシアニンなど、1 種類の色素材料を異なる金属電極の間に挟み込んだ構造であった(図 3)。しかしこれはホールと電子が同一の層を流れるために逆電子移動が起こりやすく PCE はそれぞれ 0.001 %、0.01 %と低いものであった 6, 7)。
そこで p 型材料(銅フタロシアニン)と n 型材料(PTCBI)を層状に重ねた pn 構造が Tang らにより報告された 8)。この構造では、 p 層と n 層の界面でホールと電子のペアであるエキシトンが形成され、ドナーの LUMO からアクセプターの LUMO への光励起電子移動が起こり、電荷が生じる。ホールは p 層を、電子は n 層を通ってそれぞれ電極へと運ばれる。 Tang らが報告した最初の pn 構造では PCE は 0.95%、 FF は 0.65 まで向上した。しかし有機半導体の励起子拡散長はシリコン半導体に比べて短く 20 nm 以下である。従って薄膜が厚いと励起された分子が拡散して pn 接合界面で電荷分離する前に基底状態に戻ってしまい、PCE の効率 が上がらない。一方、薄膜を薄くすると光の吸収効率が下がる。その解決法の一つとしては pn 層と pn 層の間に薄い電極を挟みこむタンデム構造がある 9)。タンデム構造にすることで、膜厚が薄い 2 つ以上のデバイスを直列に連結することができ、全体的には膜厚を厚くすることが可能になり吸光度を稼ぐことができる。また2つの層の吸収波長領域をずらし、幅広い太陽光を利用することも可能になる。
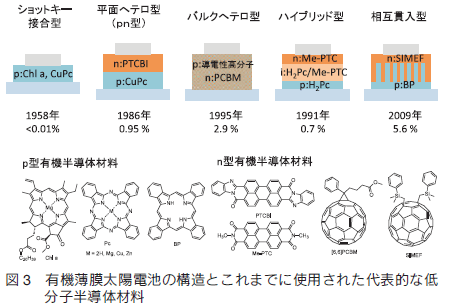
もう一つの解決法は、バルクへテロ構造である。バルクへテロ構造とは、 p 層と n 層が混じりあった構造であり、高分子薄膜太陽電池で主流となっている方法である 10, 11)。溶媒に可溶な p 型材料と n 型材料を適当な割合でブレンドし、溶液プロセスにより薄膜化する。これにより、任意の割合で p 型材料と n 型材料を混合できるだけでなく、 p 層と n 層の界面面積が大きく増加するため、励起子拡散距離が短くても、電荷分離の効率を高めることができる。このバルクヘテロ構造は、有機薄膜太陽電池において大きなブレークスルーであり、PCE が大きく増加する要因となった。さらに、 p 層と n 層の間に混合層( i 層)を挟み込んだ pin 構造も報告されている。
低分子有機半導体でこのバルクへテロ構造(ここでは pin 構造の i 層も含む)を構築するには 2 つの手法が報告されている。共蒸着と溶液塗布である。共蒸着法とは、 p 型材料と n 型材料を同時に蒸着し、結晶性の混合薄膜を作成する方法である 12)。高い半導体特性を持ちながら難溶性の、フタロシアニン、ペンタセンなどの p 型材料とフラーレンやペリレンビスイミドなどの n 型材料の利用に適した方法である。平本らは共蒸着に用いるフラーレンを高純度化することで膜厚を 1μm にすることに成功し、H2PC / C60 系で PCE = 5.3 %を達成した13)。一方溶液塗布法は、 p 型及び n 型の可溶性の低分子材料を混合し、スピンコート法などで薄膜化する方法である。近年はローバンドギャップポリマーからヒントを得た化合物や、アセンやフタロシアニンに置換基を導入して可溶化した化合物、液晶性の化合物など様々な可溶性低分子系の材料が報告されている 1, 2)。
低分子を可溶化させるもう一つの手法として前駆体法があげられる。フタロシアニンに良く似た構造のベンゾポルフィリン(BP)は、難溶で合成と精製が難しい化合物であった。しかし可溶性のビシクロ [2.2.2] オクタジエン縮環前駆体(CP)を精製した後、固体やフィルムのまま加熱し逆 Diels-Alder 反応により結晶性有機半導体のBPへと変換できることが報告された 14)。2009年には BP とフラーレン誘導体 (C60 (CH2SiMe2Ph)2: SIMEF)を組み合わせた熱変換型前駆体法による有機薄膜太陽電池が報告され、低分子塗布型材料を用いた素子としてはきわめて高い値 5.6 %のエネルギー変換効率が達成された 15)。この手法では溶液塗布でありながら、BP と SIMEF の結晶性が高く、直径 25 nm 程度の柱が櫛状に組合わさった相互貫入型の構造となっていることがポイントであった。
4. 光変換型前駆体法によるアセンの合成
我々は、このようなバルクへテロ構造あるいは相互貫入型構造を光を利用して作ることを念頭に材料の開発を行っている。熱変換型前駆体法に対し光変換型前駆体法は、光のエネルギーを利用した脱離反応であり、光を利用するため局所的な変換が可能になり、光によるプロセス制御が期待される。変換後に結晶成長を促進するためのアニーリングは必要であるものの、光反応自体は室温またはそれ以下で進行するため、デバイス作製過程で熱変換前駆体法ほど高い温度を必要としない。従って、ポリマー中に混ぜたりフレキシブル基板などの上にデバイス作製することも期待できる、新しい手法である。本章では、最近我々が開発した材料を中心に紹介する。
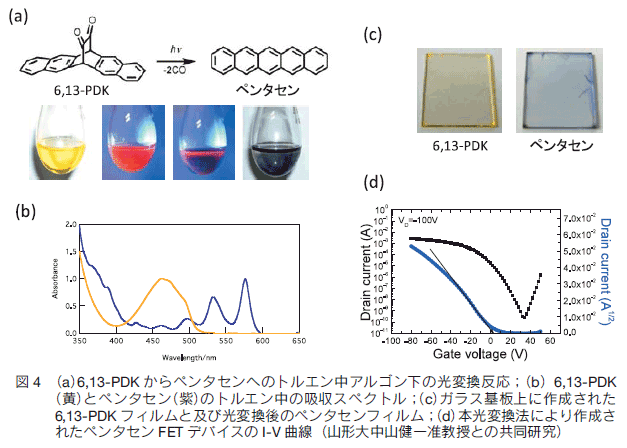
4.1 6,13- ペンタセンジケトン前駆体の光変換と塗布型ペンタセンFETの作成
我々は光変換型前駆体法を優れた有機半導体材料であるペンタセンに応用し、2005 年にペンタセンジケトン前駆体(6,13-PDK)を報告した(図 4a)。熱変換型の前駆体はそれまでにいくつか報告されていたが、光変換型は初めてであった。本章ではその光化学的挙動について述べる。6,13-PDK は、熱的には極めて安定であり、300℃ 以上に温度を上げても分解が見られず、昇華精製が可能である。図 4b に吸収スペクトルを示す。460 nm 前後に n-π*吸収が観測されるが、その吸光係数は 1400 と、カルボニル基の n-π*励起としてはかなり大きい。通常は禁制遷移であるカルボニル基の吸収の禁制がかなり緩和されて許容遷移になっているためである。
トルエン溶液中で 6,13-PDK の n-π*吸収を励起すると、2 分子の一酸化炭素が脱離し、ペンタセンへと定量的に変換する。ペンタセンのトルエンへの溶解度が低いために、反応中にペンタセンの結晶が析出する(図 4a)。この光反応の量子収率は化学光量計により 0.014 と求められた。また、本光反応は一般に紫外部のπ-π*励起でも進行する。
本光反応はフィルム中も進行する(図 4c)。この 6,13-PDK を用いた塗布型有機薄膜トランジスタの作製を山形大中山健一准教授との共同研究により試みた。ペンタセンはこの n-π*吸収はちょうど青色 LED 発光波長に相当する。スピンコート法による前駆体の塗布と電界効果移動度は最大 0.86 cm2/Vs を記録した(図 4d) 16)。これは、一般的なペンタセン蒸着膜による FET 特性に匹敵する値であり、この値が溶液塗布プロセスから成膜した薄膜で観測されることは注目に値する。
また我々は 6,13-PDK の構造異性体である 5,14-PDK の合成に成功した(図 5) 17)。興味深いことに 6,13-PDK とは吸収スペクトルの形が大きく異なる。通常の n-π*吸収に加えアントラセンのπ-π*吸収に由来する吸収が観測されるが、さらに 400 nm 付近にブロードな吸収が見られる。400 nm はちょうど生成物のペンタセンの吸収の谷間に相当し、また赤色 LED の発光波長に相当する。 5,14-PDK のトルエン中での光反応量子収率は、400 nm,460 nm のいずれも 6,13-PDK を超える 0.024 であり、より広範囲の光の利用が可能となった。
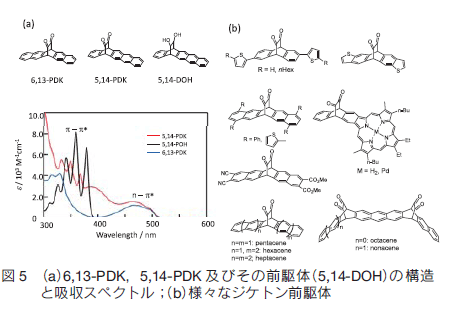
4.2 様々なジケトン前駆体
本反応はアントラセン系有機半導体材料やペンタセン誘導体、高次アセンやポルフィリン系にも展開可能である(図 5b)。アントラセン系化合物では、n-π*吸収と光変換後のアントラセンの π-π*吸収が重ならないために、大気安定性が高く、またフィルター効果も小さいために光反応が早く進行するという利点がある 18)。太陽電池の材料として用いるには、吸収波長領域の点で劣るが、 OFET などの用途には優れている。また、ペンタセンは難溶性であると同時に大気下で不安定なために、置換基を導入する位置が比較的限られているが、ジケトン部位を保護基として利用することで、 6,13 位の最も反応性の高い部分を保護し、同時に溶解度も上げて置換基導入を行うことができる。最終的に光変換を行うことで、 6,13 位は無置換のまま別の部位に置換基が導入された置換ペンタセンの合成が可能となった 19, 20, 21)。一方前駆体のベンゼン環が 6 以上連結したアセン類は、溶解度と酸素安定性が極めて低いために合成が困難とされていたが、本光反応を利用することで可能となった。最終段階で高次アセンへと変換でき、しかも低温、フィルム中、気相反応での変換が可能なために、通常の方法では合成が困難な化合物の合成が可能である。これまでに本光反応を利用したヘキサセン、ヘプタセン、オクタセン、ノナセンの合成が報告された 22-25)。一方我々はモノアンスラポルフィリンのジケトン前駆体の Soret 帯をトルエン中で励起することでモノアンスラポルフィリンへと変換することにも成功した 26)。
4.3 新しいペンタセン誘導体の開発
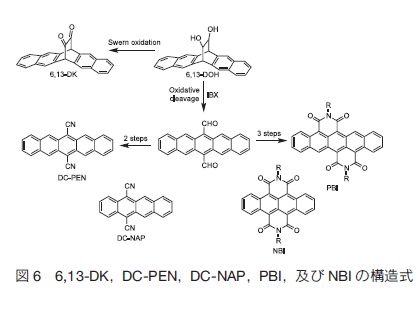
最後に 6,13-DK の合成中間体を利用することで合成が可能になったペンタセン誘導体を紹介する。6,13-DK はジヒロドキシ誘導体(6,13-DOH)を Swern 酸化することにより得られるが、6,13-DOH を 2- ヨードキシ安息香酸(IBX)により処理すると、酸化的に開裂し、6,13- ジホルミルペンタセンが得られることを見いだした。この化合物はペンタセン誘導体合成の新しいキー化合物として利用できる。例えば、このジホルミルペンタセンを出発物質に、ジシアノペンタセン(DC-PEN)やペンタセンビスイミド(PBI)の合成に成功した。これらはいずれも新規化合物であり、同様の合成法でジシアノナフタセン(DC-NAP)、ナフタセンビスイミド(NBI)も合成した。ジシアノアセンは強い蛍光を示し、またアンビポーラー特性を示すことがわかった 27)。一方テトラセンビスイミドは n 型 FET 特性を示した 28)。
5.終わりに
6,13-PDK を用いた光変換有機半導体材料は、光を用いることで、光の強度、照射時間、波長など、多くのパラメータで制御することが期待される。またレーザー技術を用いることで、2 次元のみならず 3 次元のパターンニングも可能である。 6,13-PDK は熱には極めて安定であるため、光の自由度を最大限に利用した薄膜構造制御が期待できる。光変換前駆体法で作成した薄膜で蒸着膜に匹敵する FET 特性が実現されており、光制御により結晶性薄膜を作成することに成功した。この手法を展開し、光による分子レベルのプロセス制御を通じて、新しいバルクへテロ構造制御へと展開したい、と考えている。
6.謝辞
本研究の一部は、JST CREST「革新的塗布型材料による有機薄膜太陽電池の構築」における、山形大学大学院理工学研究科 中山健一准教授、千葉大学大学院理工学研究科 矢貝史樹准教授、新潟大学大学院理工学研究科 生駒忠昭准教授、関西学院大学理工学部 増尾貞弘准教授との共同研究によるものです。また、本研究を推進するにあたり愛媛大学大学院理工学研究科、小野昇名誉教授、宇野英満教授、奥島鉄雄准教授、愛媛大学総合科学研究支援センター森重樹特任講師をはじめ多くの共同研究者、愛媛大学大学院理工学研究科有機化学研究室及び奈良先端科学技術大学院大学物質創成科学研究科有機光分子科学研究室の学生諸氏のご協力を得た事を感謝致します。本研究の一部は文部科学省科学研究費(B)及び特別経費「グリーンフォトニクス研究教育推進拠点整備事業」の支援により行われました。
| 著者プロフィール | |
| 氏名 | 山田 容子 (Hiroko Yamada) |
|---|---|
| 所属 | 奈良先端科学技術大学院大学 物質創成科学研究科 教授 |
| 連絡先 | 〒630-0192 奈良県生駒市高山町 8916-5 TEL : 0743-72-6041 FAX : 0743-72-6042 E-mail :hyamada@ms.naist.jp |
| 略歴 | 1992 京都大学大学院理学研究科化学専攻 博士後期課程修了・博士 (理学) 1992-1994 日本学術振興会特別研究員 (PD) 1994-1998 ㈱日本チバガイギー国際科学研究所及び㈱チバ・スペシャルティー・ケミカルズ 1998-2000 大阪大学産業科学研究所機関研究員 2000-2003 JST CREST 博士研究員 2003-2010 愛媛大学理学部助教授(2007 より准教授に名称変更) 2011-2012 奈良先端科学技術大学院大学 物質創成科学研究科准教授 2012- 現職 2006-2010 JST さきがけ「物質と光作用」領域研究員(兼担) 2010- JST CREST「太陽光エネルギー」領域研究代表(兼担) |
| 専門 | 有機化学,有機光化学 |
| Copyright(c) 1996-2012 DOJINDO LABORATORIES, ALL Rights Reserved. |